
About us
We are a medicinal chemistry research group based in the School of Pharmacy at the University of East Anglia, Norwich, UK. We focus on the development of new therapeutics, with a particular interest in targeting DNA and protein-protein interactions.
The group uses both solution and solid phase synthesis to design compounds derived from natural products or purely synthetic sources.
Some highlights include: in 2007, in collaboration with Christine Cardin, we described a new mode of binding to DNA that targeted a four way junction called a Holliday Junction (HJ). In 2010, we showed that we could induce the formation of a HJ at room temperature, another first with potential applications in nanotechnology as well as therapeutics. In 2003, we carried out the first solid phase synthesis of the chlorofusin peptide, part of a natural product targeted at the p53/MDM2 interaction and followed it with the solid phase synthesis of a triostin A analogue that represented the most efficient synthesis of this DNA binding molecule to date. We are currently focussed on targeting protein-protein interactions and DNA targets in cancer and inflammation, alongside the synthesis of natural products targets such as simocyclinone.

Targeting DNA
One of the main focuses of the laboratory is the targeting of DNA as a potential route to therapeutically useful molecules. Many clinically used antitumour agents depend on the disruption of nucleic acid associated processes to exert their effects. In the future, new agents that have lower side effects and are more focussed on single targets (either genes or structures) will be required and this is a subject of investigation.
We are interested in both synthetic molecules and natural products and combine studies of synthesis, biophysical and biochemical properties. In 2006, in collaboration with Christine Cardin at Reading University, we disclosed the first structure of a molecule binding to the four-way Holliday junction through a novel mode of action. In 2011, we followed this with the first molecule that can promote the assembly of the junction at room temperature. This compound was derived from a click chemistry approach that led to a series of compounds that could bind to various DNA structures, from duplexes to G-quadruplexes via junctions.

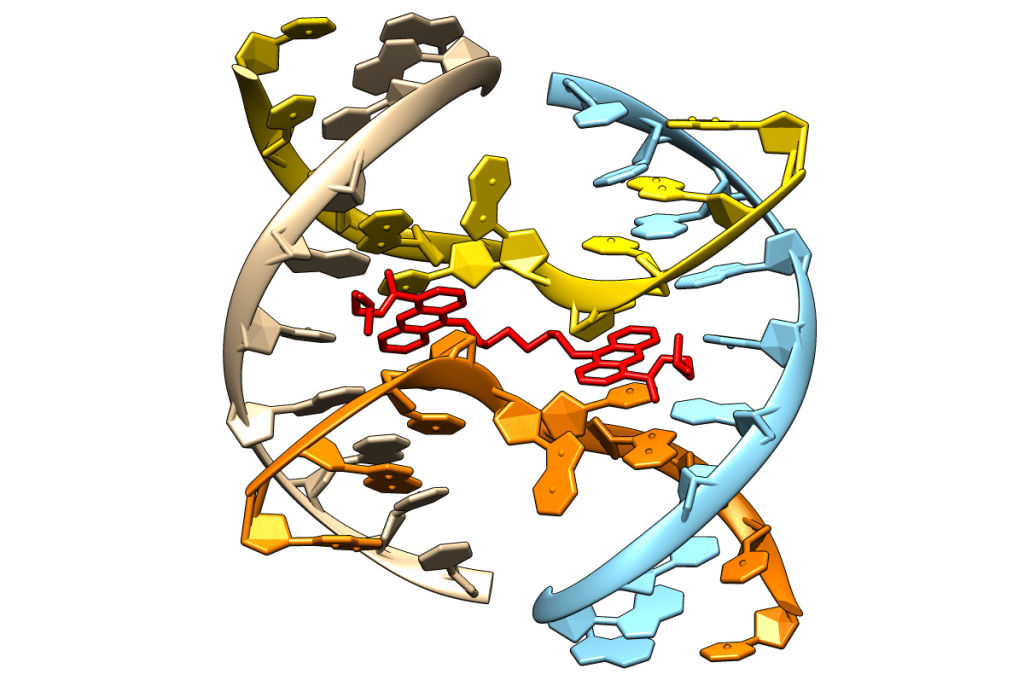
Molecules that bind to double stranded DNA still have potential as antitumour agents. With Laurence Patterson of the Bradford Institute for Cancer Therapeutics, Searcey proposed that the duocarmycins, ultrapotent antitumour natural products, could be redesigned to become prodrugs that were activated by bio-oxidative processes in cancer cells. Working with Klaus Pors and the Bradford team, we have demonstrated that this is indeed the case and have shown that the prodrugs have antitumour activity in cells expressing cytochrome P450 enzymes.

Targeting of ultrapotent antitumour agents, most notably with antibodies to form antibody-drug conjugates, is fast becoming one of the clinical approaches of choice in oncology. In 2015 we disclosed the first synthetic route for an Fmoc-protected duocarmycin alkylating unit appropriate for solid phase peptide synthesis, and have demonstrated its application. We have used both peptides and small molecules to target the duocarmycins to cancer cells and, with David Russell and Maria J. Marin, have developed gold nanoparticles with potential in photodynamic therapy.

Protein-protein interactions
Signalling pathways in cellular systems are excellent targets for drug design. The plethora of projects focussed on designing kinase inhibitors comes from this recognition and the progression of imatinib into the marketplace. Whereas kinases have a defined binding site, signalling can also take place through two protein surfaces coming together, a daunting prospect for the design of inhibitors but one which is coming to the fore in the design of new agents. In 2003, we described the first synthesis of the peptide structure of chlorofusin, a natural product that has been shown to inhibit the interaction between p53 and MDM2. The peptide lacked inhibitory activity, as did a series of analogues and later work showed that the full structure of the natural product is required for activity, although the stereochemistry of the azaphilone was relatively unimportant.
In a collaboration with the O’Connell group at UEA, we have studied the Nrf2/Keap1 interaction. Inhibition of this interaction has potential in cancer chemoprevention and in inflammation and in 2013, we disclosed the first cell penetrating peptide to activate Nrf2 through binding to Keap1 and also small cyclic peptides. We are currently investigating small molecules that may also have an effect. We have developed a number of in vitro and cell culture assays in house in order to do this. Ironically, Nrf2 upregulation may also be a problem in cancer and we are also investigating Nrf2 as a target for inhibition.
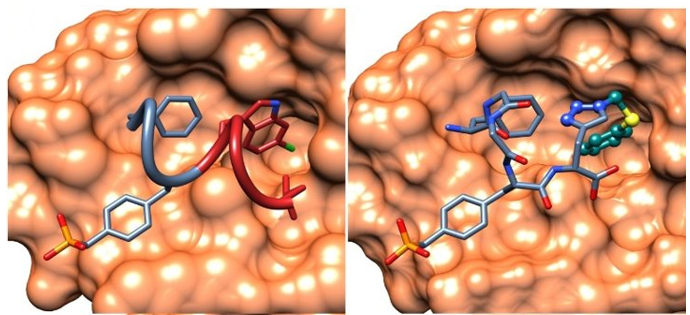
Protein-protein interactions often have flat, undefined surfaces and are hard to target using small molecules. We have developed a method called Peptide Directed Ligand Design, working with the Beekman group and Lesley Howell (QMUL), to design and synthesise small molecules targeting these difficult interactions. In our initial studies, we have seen excellent success rates where 50% of molecules designed and synthesised bind to their target protein.

Synthesis and natural products
At the centre of all of our projects is organic synthesis. We use both solid and solution phase methodologies to generate the structures that are of interest to us. Often, this may involve the development of routes to natural products in order to study structure-activity relationships. In 2005, following on from our successful solid phase synthesis of the chlorofusin peptide, we synthesised the triostin A analogue TANDEM via a route that also allowed us to generate water soluble analogues. We are applying solid phase methodology to the synthesis of other natural products.
Heterocyclic chemistry sits at the heart of most of our DNA targeting, with routes to acridines and analogues via, amongst others, classical and benzyne click chemistry, well established. We are currently investigating DNA gyrase inhibitors via the development of flexible routes to coumarins and analogues.



